





Children De JAH
TAFARI GENESIS
RETREAT CAMP
West Africa
Tafari : (Amharic) to be feared, revered. Genesis : creation beginning.
Children with life-threatening conditions are forced to grow up too fast.
We particularly choose children whose lives have been affected by civil wars,
Former Child Soilders, or who suffers chronic life threatening illnesses such as
Sickle Cell Anemia, Malaria, Asthma, Aids, all of which can be a death sentence
in Africa and Haiti.
- We provide counseling,
- teach various coping and meditation techniques to deal with physical pain crisis.
- We address their needs at home, and follow up on their progress.
- Malaria kills more children than Aids. We support existing mosquito net programs which educates families about simple steps to help prevent malaria, and provides low cost or free nets to mothers. A child with an existing life threatening illness has little chance to survive once they get malaria. Many mothers must make the choice of food, medicine or mosquito nets. We don’t think they should have to make such a choice.
- We supply JAH Glory Knapsack's with books, music, and much needed school
- supplies and art materials to all kids.
TO DONATE "God is Our Hope " - Alpha Blondy-
Inicheh / Merci / Thank You for your kind support.
ALPHA BLONDY JAH GLORY FOUNDATION©
Benefiting African Women and Children Charities in Africa and Haiti
Oganisation charitable femmes et enfants Africains et Haiti
Site official / Official Site of Alpha Blondy Jah Glory Foundation. Inicheh / Merci / Thank You for your kind support.


WELCOME ". . .and Babylon shall not rise again. . ." - ALPHA BLONDY -





A Sanctuary For Children

Tafari - Genesis Retreat Camp is a Sanctuary for Children. We hope to bring joy and a sense of peace and pride to the children through various activities, such as the “get to know Africa” program.
We are passionate about introducing them to the immense beauty and history of Africa. Many African children dream only of Europe or America and never learn about the vast beauty and great history of their homeland, because all they know is pain, suffering and death. We aim to heal the precious souls of the children. We aim to provide a safe place where they are allowed to just be children and not think about adult duties or conflicts, or death, even if just for a short time.
We strive to give them good memories to last a lifetime.
It should not hurt to be a child.
Click on image to enlarge


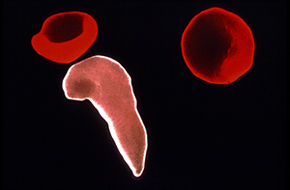
Sickle cell disease is a hereditary disorder caused by a defect in the hemoglobin gene of the red blood cells.
Fact: We need blood to live. The main purpose of red blood cells is to deliver oxygen throughout the body.
Blood circulating through the body delivers oxygen and nutrients to the cells and tissues of the body.
The blood travels throughout the body via a vast network of tube-like blood vessels, which become
smaller and smaller as they reach a hungry cell.
red blood cells called hemoglobin; it makes our blood red. This protein is responsible for carrying
oxygen you breathe in your lungs, to your heart, and out to the rest of your body - eventually realeasing it to your hungry cells.
One little change in your hemoglobin can change the shape of your blood cells; when you have sickle hemoglobin, it forms long rods
when it gives up oxygen. The genes you inherit from your mother and father determine what type of hemoglobin you make.
or the sickles that farmers use to cut tall grass (hence the name).
red blood cells go through the small artreries, they block flow, then break apart too quickly.
Anemia can result from a variety of reasons, but in a person with sickle cell disease, anemia usually occurs because the red blood cells can not be replaced as fast as they are destroyed.
Anemia can cause fatigue (tiredness) and may result in an inability to participate in strenuous activities for a long period of time.
Normal red blood cells last 90-120 days. (3-4 months).
Sickle red blood cells last 10-20 days.
Yes, there are different types of sickle cell disease, all of them named for the "shape changeing" hemoglobin (Hb).
Hb SS, (called sickle cell anemia)
Hb SC,
Sickle Beta Thalassemia
Hb SD-Punjab
Hb SO-Arab
Hb SE
Hb S-HPFH (hereditary persistence of fetal hemoglobin)
All of these combinations have sickle complications, but some are more severe than others.
There are many genetic differences within these hemoglobin combinations that are not yet fully
understood that make some have a milder course and some have a more severe course and a shorter life span.
Sickle Beta Thalassemia
Is an inherited disorder of beta globin production, a part of the hemoglobin molecule.
Decreased hemoglobin production causes smaller than normal red blood cells, which
look like bull's eye targets under a microscope.
Beta thalassemia can combine with the sickle hemoglobin to cause sickle beta thalassemia.
Most people with beta thalassemia have fewer problems with infections, fewer pain episodes,
and less damage to the organs early in life. They often do vey well when they get older.
No, Sickle Cell is found in many other nationalities and races, including Arabs, Greeks, Italians, Latin Americans, and Indians.
But Sickle cell is most prevalent in people of African decent and Latinos/Hispanics.
Males and females have different life spans and this is true for people with sickle cell disease as well.
Some people with sickle cell disease live longer than the average life span and some people live less than the average life span.
People with different types of sickle cell disease have different average life spans and different complications from the disease.
> A female with Hemoglobin SS disease will live an average of mid 30's to mid 40's.
> A male with Hemoglobin SS disease will live an average of mid 30's to 42 years.
> A female with Hemoglobin SC disease will live an average of 68 years.
> A male with Hemoglobin SC disease will live an average of 60 years.
Related Facts:
People with Sickle Beta Zero Thalassemia have similar complications as those with homozygous sickle cell disease (SS),
so their average life spans are the same.
You are probably already aware of the dangers of alcohol, smoking and using "street" drugs.
These are harmful to everyone. But for people with sickle cell disease,
even occasional drinking, smoking (even second hand smoke) or drug use can lead to severe sickle cell complications.
Alcohol + fluid loss + dehydration = PAIN!
Smoking = less oxygen to lungs = lung infection and permanent lung damage!
"Street" drugs = serious stress to body = damage to major body organs – heart, lungs and brain!
You are in control over what you put in your body. Please choose wisely.
Yes, A Bone Marrow Transplant is the only cure for
Sickle Cell Anemia. However this cure is not available or practical for
most sicklers due to the risk of rejection.
A sibling who closely matches the patient or a stem cell transplant provides
the best chance for a successful transplant, because the posibility of rejection
by the patient's body will be greatly reduced.
Umbilical cord blood
stem cell transplants
are less prone to
rejection.
(A bone marrow transplant using stem cells taken from cord blood.)
How do alcohol, "street" drugs and tobacco affect people with sickle cell disease?
Are there different types of sickle cell disease?
How long can people with sickle cell disease live?


Healthy red blood cell
Sickled red blood cell
Hemoglobin SS - Sickle Cell Anemia
is caused by inheriting two sickle genes, one from each parent. This is the most severe
form of the Hb group with the shortest life span.
Symptoms may include moderate to severe anemia, increased infections, tissue damage,
organ damage, mild to severe stokes because of blocked blood flow to the brain, blockage of the
eye blood vessels may lead to loss of vision. As patients age, damage to lungs and kidneys can
cause breathing problems.
Severe pain episodes can be unpredictable and attack any part of the body and often require
hospitilization, and can be disruptive to the normal life of the person and their family.
Treatment for pain crisis often includes narcotics such as morphine given intraveniously,
blood transfusion, and oxygen to replenish the blood.
Hemoglobin SC
people with Hb SC - sickle cell , inherit a Hb C gene from one parent and a Hb S gene from the other. In General, those with Hb SC have a syndrome very similar to sickle cell anemia, though the hemoysis is usually less severe, so the anemia is milder.
The reported average life expectancy of those with Hb SC is in the mid -60's compared to
the mid 30's to 40's of those with Hb SS.
There may be more eye problems, bone damage, such as AVN, or vascular necrosis,
of the hip and shoulder bones, is more common, so chronic pain may become a problem
in older patients.
Hb SD-Punjab, Hb SO-Arab, Hb SE,
These are milder forms of sickle cell disease with occassional pain episodes.
Hb S-HPFH,
This form of sickle cell disease is rare with only five reported cases. These cases were all mild.
The Hb E gene is very common in many areas of Southeast Asia, India, and China.
Is Sickle Cell Anemia Disease found only in people of African desent ?
QUESTIONS ABOUT:
QUESTIONS SUR:
According to the World Health Organization (WHO).
Of all child deaths worldwide, Africa had 94% of those caused by malaria deaths, 89% from HIV/AIDS,
46% due to pneumonia, 40% from diarrhea, and 5% from measles, says the WHO.
The numbers are based on published or publicly available records. Those aren't perfect, the report notes.
Selon l'Organisation mondiale de la santé (OMS).
De tous les décès d'enfants du monde entier, l'Afrique avait 94% de ces décès causés par le paludisme,
89% du VIH / sida, 46% dus à la pneumonie, la diarrhée de 40%, et 5% de la rougeole, a déclaré l'OMS.
Les chiffres sont basés sur publiés ou rendus accessibles au public. Ceux qui ne sont pas parfaits, indique le rapport.
Finding ways to distract from the pain,
such as yoga, meditation, music, reading,
or in this case "texting" can be helpfull.
At the TAFARI GENESIS RETREAT for Children in West Africa, we have volunteers who teach
arts and crafts and various meditation techniques. Many African children do not have access
to pain medication or hospital care, so learning ways to cope with chronic pain is essential.
Genesis herself helps the younger children with various ways to deal with pain,
using her own experience as example.
Is there a cure for Sickle Cell Anemia Disease ?
Malaria is an infection of the blood that is carried from person to person by mosquitoes.
The disease has been recognized for thousands of years and once was found almost everywhere except in the most northern areas of the world. Malaria has been wiped out in North America, Western Europe, and Russia. It remains a serious problem in much of the tropical and subtropical world, however.
Millions of people continue to be infected every year, and probably up to 1 million of them die.
Le paludisme est une infection du sang qui est transmis d'une personne à une autre par les moustiques.
La maladie a été reconnu depuis des milliers d'années et une fois a été retrouvé à peu près partout sauf dans les zones les plus septentrionales du monde. Le paludisme a été anéantie en Amérique du Nord, l'Europe occidentale et la Russie. Il reste un sérieux problème dans une grande partie du monde tropical et subtropical, cependant.
Des millions de personnes continuent à être infectées chaque année, et probablement jusqu'à 1 million d'entre eux meurent.
By the time you read this 2 children will die from malaria
Getting IV pain medication
Photo P. Whyte ©2006
What is Sickle Cell Anemia Disease ?
Infection Threat
The major threat to children comes from massive infection.
They start out, they have a little fever and get a little cranky. Most kids, you give them tylenol, you put them to bed and they get better, but kids with sickle cell, they just get worse and worse. They just crash !
~~~~~~~~~~~~~~~~~
They go from zero to
death's door in minutes
and you will never
know just by looking at
them, how fragile they
can be.
" The pain inside my bones
feels like it's the worst pain
you can imagine multiply by
a million.
I wonder why God has to make
it so bad.
Sometimes even the morphine don't help"
"When the pain is in my side or back it's like getting stabbed a
million times very fast. In my
lungs, it hurts to even breathe.
You're just tired of all this pain,
and you know that it will come again and again and again"
- GENESIS.
You have to be strong, because when it's your children, you have nothing if you don't have strength and hope.
Your child needs you to fight for them.
A low-grade cold can be a harbinger of swift death.
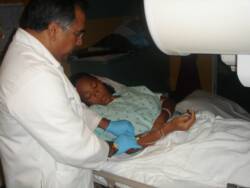
TAFARI who has sickle cell trait comforts his sister
GENESIS (10 yrs old) who has sickle cell anemiaHbSS;
as she receives a blood transfusion.
Where there is ignorance, there's myth, and myth's surround sickle cell disease. Health education is about dispelling
health myths such as :
- Sickle Cell Disease is not contagious. You cannot catch it from someone; it is a genetic disorder you receive from birth.
- It is not a cancer
- It is not a "black" disease, but affects Hispanics and people of Asian, and Mediterranean origin as well.
- It is not "bad" blood
- The mind is not affected. Children with sickle cell disease sometimes do poorly in school because of poor attendance due to illness and they usually must work twice as hard in school to keep up. They are usually quite brilliant in this endeavor.
- It is not a curse, voodoo, or a spell that someone cast .
- You cannot "grow out of it". . . it is life long.
Sickle Cell Disease is a
genetic disorder inherited from
two parents who each have the
sickle cell trait.
A child conceived by two people
with the trait has a 1 in 2 chance of
having the trait; and a 1 in 4 chance
of having sickle cell disease.
Researchers believe the mutation
happened as a way for the body to
protect itself from Malaria. People
with sickle cell has immunity
from Malaria.
Disease, however it is more poison
than protector. They are not
protected from Malaria....It is pain
as birthright.
Misery mapped onto their genes.

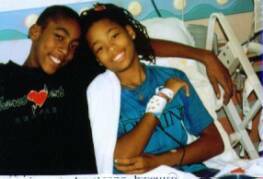
Genesis (22 yrs old) "chillin" and texting friends.
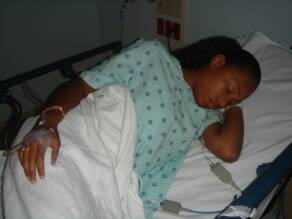
Sicklers sometimes are forced to stay in bed for days
taking strong pain medication due to a sickle cell pain crisis.
Radioacive dyes being injected in IV to get a
scan of internal organs to check for damage
from sickle cell crisis. Photo P. Whyte ©
In 2006 the US postal service
issued an official stamp to bring
awareness to Sickle Cell Anemia
please visit the official SC site at
How do you get Malaria ?
Comment obtenez-vous le paludisme?
Mosquito nets that are treated with insecticide and used properly each night has been proven to
greatly reduce the high rate of malaria in children.
Que les moustiquaires traitées avec des insecticides sont utilisés correctement et chaque nuit a été prouvé
que de réduire considérablement le taux élevé de paludisme chez les enfants.
We support existing mosquito net programs that meet our criteria for helping
women and children.
We specifically target mothers with children who are fighting chronic illnesses,
to educate them on the importance and proper ways to use the nets.
For a child who already has a chronic illness, malaria can mean a swift death.
Many mothers must make the choice of food, medicine or mosquito nets.
Pour un enfant qui a déjà une maladie chronique, le paludisme peut signifier une mort rapide.
Beaucoup de mères doivent faire le choix de la nourriture, des médicaments ou des moustiquaires.
We don’t think they should have to make such a choice.
Nous ne pensons pas qu'ils devraient avoir à faire un tel choix.
With your donations, you can help us get more of these nets
into the hands of mothers to help them protect their children.
" God is Our Hope " - ALPHA BLONDY -
Simple ways to protect children from Malaria death
Trucs pour protéger les enfants contre le paludisme mort

Former
CHILD SOILDERS
It is often said that the eyes
are the windows to the soul.
When you look into the eyes of these children it is difficult to see
the light...still they shine bright inspite of the evil they have had to
endure in their young lives.
Child Soilders are not killers by their own choice.
These are innocent children that were violently kidnapped from
their homes, some as young as 4 years old, and forced to carry
machetes and guns and to kill or be killed.
Many children watched as their parents and other family members
were raped and murdured. Some of the bodies were cut up in
pieces and cooked in a huge pot, which the children was then
forced to eat, while a gun was being held to their heads.
Horrible, and unimaginable.
This was just a small part of the mind control that was used to
force these innocent children to become killers.
Children were given drugs to keep them high and make them feel
that were invincible so they could in turn carry out more raids on
other villages and kidnap and murder others, just like themselves.
One child said, when he was given "the white thing" he felt numb,
like he could do anything and feel nothing.
(It has been reported that they were given cocaine and also
a certain kind of leaf to chew, found only in the jungle, which
gives a narcotic effect.)

Former Child soilders in a Uganda Orphanage
singing a song for peace and no more wars.
These children were rescued from northern Uganda.
The young girls had been raped and the young boys abused.

Photo Douna Clarke ©2006
Photo P. Whyte ©2006
Photo P. Whyte ©2003
Photo Douna Clarke ©2006
Photo P. Whyte ©
~ MALARIA IN CHILDREN
Paludisme chez les enfants
Each night in northern Uganda, tens of thousands of terrified
children leave their villages at dusk and walk miles to town to
avoid being kidnapped by the Lord's Resistance Army—a brutal
rebel force that has abducted more than 30,000 children to
serve as soldiers and slaves in its 20-year war against the
Ugandan government.
They are known as "The Children of the Night", they repeat this
journey each and every night to sleep on the ground and then
walk back the same miles to their village, without even a small
breakfast. Still they count themselves lucky to be alive and free.
Any size donation does matter and each dollar
goes towards education and health programs.
We give childrens books, school supplies, and art
materials to all kids to help them heal through
Art Therapy. Give Thanks for your kindness.
The boys and girls at this Orphanage in Uganda are very happy
to be free and safe, however they sleep on the floor with no mats.
The J A H Glory Foundation is engaged in helping to provide
some basic human necessities for them, as well as support other
ongoing programs at various Orphanages housing former child
soilders.
" God is Our Hope "
- ALPHA BLONDY -
This is not ficton or a Hollywood movie that you watch and feel sad and cry and forget about.
It is not something that happened a long time ago. . .It is happening now. It is happening on your watch
Ce n'est pas ficton Hollywood ou un film que vous regardez et se sentent tristes et pleurer et oublier.
Ce n'est pas quelque chose qui s'est passé il ya longtemps. . . Ce qui se passe maintenant.
Elle se produit sur votre montre
Qu'il n'y ait NO MORE ENFANT SOILDERS.
young girls going to sell food
in Burkina Faso, W. Africa
Photo P. Whyte ©2004
Related Links:
Let this year count for something
THE ALPHA BLONDY J A H GLORY FOUNDATION
supports the existing grassroots programs in these areas, mainly
run by mothers who are trying to keep the children safe.
Douna Clarke, our traveling nurse, volunteers to help feed
and vaccinate the children at the Orphanage.
What J A H Glory is doing to help save the lives of children
Malaria is spread when an infected Anopheles mosquito bites a person.
Only this type of mosquito can spread malaria. These mosquitoes are
active from dusk to dawn.
The mosquito becomes infected by biting an infected
person and drawing blood that contains the parasite.
When that mosquito bites another person, that
person becomes infected.
Le paludisme se transmet lorsqu'un moustique anophèle
infecté Morsures d'une personne. Seul ce type de moustiques peuvent
se propager Paludisme. Ces moustiques sont actifs du crépuscule à
l'aube. Le moustique est infecté par piqué une personne infectée
Et le dessin du sang contenant le parasite. Lorsque que les piqûres
de moustiques pour une autre personne, cette personne Est infecté.
L'ALPHA BLONDY J A H GLORY FOUNDATION
Soutient la base des programmes existants dans ces
domaines, principalement dirigées par des mères qui
tentent de garder la sécurité des enfants.
Ces enfants ont été sauvés du nord de l'Ouganda. Les jeunes
filles ont été violées et les jeunes garçons victimes de sévices.


ORPHANS
LIVING WITH HIV/AIDS
Quand les caméras des médias du monde entier est passé et les yeux du monde sont détournés pour le prochain "reportage". . . Les enfants sont toujours là.
Les orphelins sont toujours victimes et leur nombre continue de faire plus grand.
Nos orphelins sont infectées par le VIH pour des raisons indépendantes de leur volonté.
Certains de nos orphelins n'ont pas le VIH, mais ils sont seuls et dans certains cas, ils prendront soin de frères et sœurs qui pourraient être infectés, tout en essayant d'aller à l'école, pour qu'ils puissent avoir une vie meilleure.
We give support to existing charities in Africa that meet our critirias. Some of the critirias includes but is not limited to, is that they be non-government, non political, non profit, and operate at a grassroot level, Person to Person.
Nous donnons un appui aux institutions de bienfaisance en Afrique qui répondent à nos critirias. Certains des critirias comprend mais n'est pas limité à, c'est qu'ils soient non gouvernemental, apolitique, sans but lucratif, et de fonctionner à un niveau de base, de personne à personne.

When the cameras of the world's media is gone and the eyes of the world is diverted to the next "news story" . . . the children are still here. The Orphans are still suffering and their numbers are still getting larger.
These Orphans are infected with HIV through no fault of their own.
Some of the Orphans do not have HIV, but they are alone and in some cases they are caring for brothers and sisters who may be infected, all the while trying to attend school, so that they may have a better life.